consensusNetR is an R Package for combining networks into a consensus network based on the work of Laura Cantini and Andrei Zinovyev. In addition to identifying consensus based on correlation of community meta-genes (loadings or membership scores), we also implement methods based on overlap.
Example Workflow
This example will create a consensus network from three public datasets: GSE39582, TCGA COAD, and TCGA READ
This example workflow begins with icWGCNA results, however alternative methods work as well (KNN, PCA, ICA, WGCNA). See Example Appendix for downloading data and icWGCNA run details and code.
Consensus construction
# Create list of community_membership object
memb_list <- list(
GSE39582 = GSE39582_icwgcna$community_membership,
READ = read_icwgcna$community_membership,
COAD = coad_icwgcna$community_membership
)
# Construct Meta Reciprocal Best Hits based on overlaps (318 communities found)
rbh <- construct_rbh_overlap_based(memb_list, top_n = 25)
nrow(rbh)
## 318
# RBH Heatmap Creation
plot_rbh(rbh = rbh, network_membership_list = memb_list)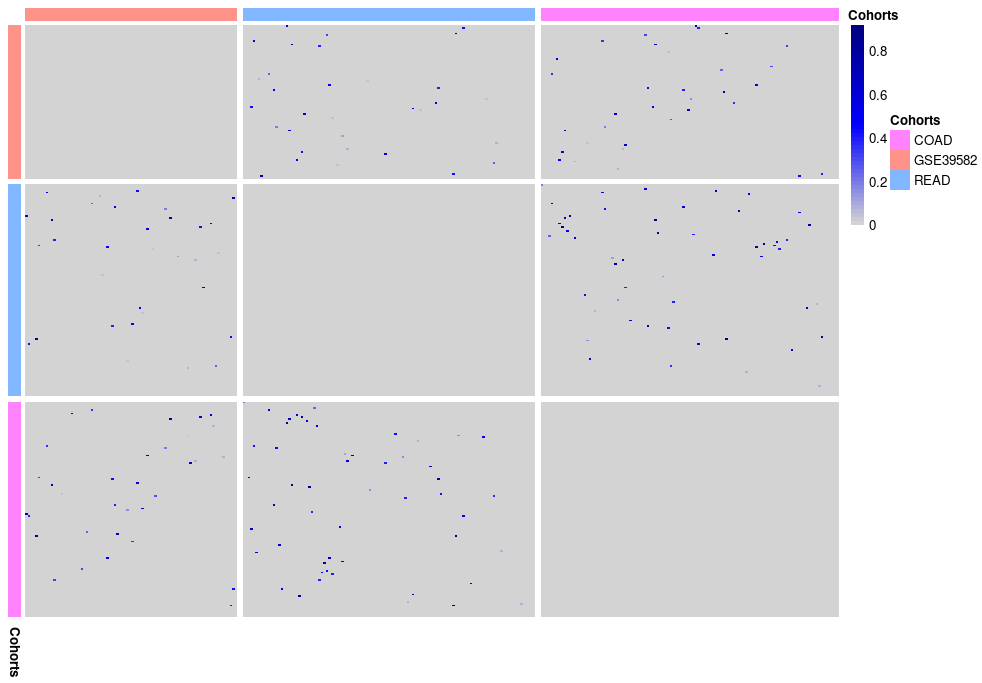
# Detect Communities in Adjacency/Reciprocal Best Hits Matrix
consensus_comms <- detect_consensus_communities(rbh)
# showing the first 10 communities
#note community 1 is a miscellaneous and will be removed
table(consensus_comms$Cluster)[1:10]
## 1 2 3 4 5 6 7 8 9 10
## 200 3 3 3 3 3 3 3 3 3
# Compute the average metagene across studies for each community
consensus_memb <- calc_consensus_memberships(consensus_comms, memb_list)Downstream Analysis
consensus_genes <- get_gene_community_membership(consensus_comms, memb_list, 2)
head(consensus_genes)
## gene_id cluster n_studies
## 1 A1BG 43 2
## 2 A2M 1 2
## 3 AACS 19 2
## 4 AAGAB 25 2
## 5 AASDH 40 2
## 6 AASS 42 2
# Need to use icWGCNA for individual eigengenes
GSE39582_eigen <- icWGCNA::compute_eigengene_matrix(
ex = GSE39582_df,
membership_matrix = consensus_memb
)
read_eigen <- icWGCNA::compute_eigengene_matrix(
ex = read_df,
membership_matrix = consensus_memb
)
coad_eigen <- icWGCNA::compute_eigengene_matrix(
ex = coad_df,
membership_matrix = consensus_memb
)
eigen_list <- list(GSE39582_eigen, read_eigen, coad_eigen)
plot_consensus_eig_dist(eigen_list)
Example Appendix
Downloading data
For GSE39582 we need to convert from Affymetrix Human Genome U133 Plus 2.0 Array to gene symbols, by using the icWGCNA::gene_mapping() function. This matches with the two TCGA datasets already in gene symbols.
library(icWGCNA)
# GSE39582
GSE39582 <- GEOquery::getGEO("GSE39582")
# TCGA READ
UCSCXenaTools::getTCGAdata(
project = "READ",
mRNASeq = TRUE,
mRNASeqType = "normalized",
clinical = TRUE,
download = TRUE,
destdir = "/MY_PATH/data/"
)
# TCGA COAD
UCSCXenaTools::getTCGAdata(
project = "COAD",
mRNASeq = TRUE,
mRNASeqType = "normalized",
clinical = TRUE,
download = TRUE,
destdir = "/MY_PATH/data/"
)Preprocessing steps
All datasets must have consistent annotation (i.e. Gene symbols, Entrez, Ensembl, …). In this example we will convert GSE39582 to gene symbols using the icWGCNA::gene_mapping() function.
# creating annotation file for gene mapping to gene symbols
GSE39582_annotation <- GSE39582@featureData@data |>
dplyr::select(ID, gene_symbol = `Gene Symbol`) |>
dplyr::mutate(
gene_symbol = purrr::map(
gene_symbol, ~ stringr::str_split(.x, " /// ")[[1]]
)
) %>%
tidyr::unnest(gene_symbol)
GSE39582_hugo <- icWGCNA::gene_mapping(
GSE39582@assayData$exprs,
GSE39582_annotation,
compress_fun = "highest_mean",
compress_trans = "log_exp"
)All data should be normalized, however in this example we downloaded the normalized data so no transformations needed here.
icWGCNA runs
For icWGCNA runs using defaults, except reducing max iterations to 5 for demonstration purposes. These runs benefit greatly by using multiple computer cores.
Code of Conduct
Please note that the icWGCNA project is released with a Contributor Code of Conduct. By contributing to this project, you agree to abide by its terms.